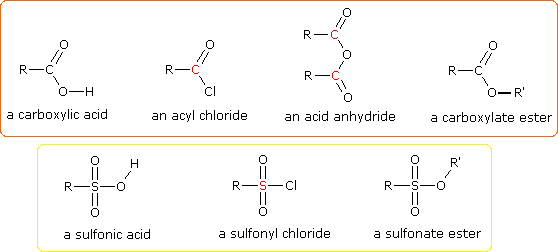
Nomenclatura degli alcoli |
|---|
Nel sistema di nomenclatura IUPAC, i gruppi funzionali vengono designati in
uno o due modi. La presenza della funzione può essere indicata da un suffisso
caratteristico o da un numero di posizione. Questo si verifica comunemente per i
doppi e tripli legami carbonio-carbonio che hanno i suffissi ene e ino,
rispettivamente. Gli alogeni, invece, non hanno un suffisso e sono nominati come
sostituenti; per esempio: (CH3)2C=CHCHClCH3 è
il 4-cloro-2-metil-2-pentene. Se hai qualche incertezza sulle regole di
nomenclatura IUPAC, dovresti rivederle ora.
Gli alcoli sono usualmente denominati con la prima procedura e sono designati
con il suffisso olo, come per l'etanolo, CH3CH2OH
(notare che non è necessario un numero di localizzazione in una catena a due
atomi di carbonio). Su catene più lunghe, la localizzazione del gruppo
ossidrile richiede una numerazione. Per esempio: (CH3)2C=CHCH(OH)CH3
è il 4-metil-3-penten-2-olo. Altri esempi di nomenclatura IUPAC sono mostrati
sotto, insieme ai nomi comuni utilizzati spesso per alcuni dei composti più
semplici. Per gli alcoli monofunzionali, questa designazione con nomi comuni
consiste nella parola alcool, seguita dalla denominazione del gruppo
alchilico. Gli alcoli possono anche essere classificati in primari, secondari
e terziari, allo stesso modo degli alogenuri alchilici. Questa
terminologia si riferisce alla sostituzione alchilica dell'atomo di carbonio che
reca con sé il gruppo ossidrile (di colore blu nell'illustrazione).
Molti gruppi funzionali hanno un suffisso caratteristico di designazione, e soltanto uno di tali suffissi (oltre che "ene" ed "ino") può essere utilizzato in un nome. Quando il gruppo funzionale ossidrile è presente con un funzione con priorità di nomenclatura più alta, esso deve essere citato e localizzato dal prefisso idrossi e da un numero appropriato. Per esempio, l'acido lattico ha il nome IUPAC: acido 2-idrossipropanoico.
Composti contenenti un gruppo funzionale C-S-H sono chiamati tioli o mercaptani. Il nome IUPAC di (CH3)3C–SH è 2-metil-2-propantiolo, comunemente denominato tert-butil mercaptano. La chimica dei tioli non sarà descritta qui, oltre al fatto che è opportuno notare che essi sono acidi di più forti, e nucleofili più potenti, degli alcoli.
Reazioni degli alcoli |
|---|
Il
gruppo funzionale degli alcoli è il gruppo ossidrile, -OH. A differenza
degli alogenuri 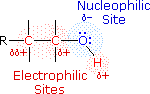 alchilici,
questo gruppo ha due legami covalenti reattivi, il legame C-O
ed il legame O-H. L'elettronegatività
dell'ossigeno è notevolmente più elevata di quella del carbonio e
dell'idrogeno. Di conseguenza, i legami covalenti di questo gruppo funzionale
sono polarizzati, per cui l'ossigeno è ricco di elettroni ed il carbonio e
l'idrogeno sono elettrofili, come mostrato nel disegno a destra. Per la
precisione, la natura bipolare del legame O-H è tale che gli alcoli sono acidi
molto più forti degli alcani (di circa 1030 volte), e quasi della
stessa misura, più forti degli eteri (alcani sostituiti dall'ossigeno, ma che
non hanno un gruppo O-H). La regione più reattiva in una molecola di alcool è
il gruppo ossidrile, a dispetto del fatto che la forza del legame O-H è
significativamente più alta dei legami C-C, C-H e C-O, sottolineando ancora una
volta la differenza tra stabilità termodinamica e stabilità chimica.
alchilici,
questo gruppo ha due legami covalenti reattivi, il legame C-O
ed il legame O-H. L'elettronegatività
dell'ossigeno è notevolmente più elevata di quella del carbonio e
dell'idrogeno. Di conseguenza, i legami covalenti di questo gruppo funzionale
sono polarizzati, per cui l'ossigeno è ricco di elettroni ed il carbonio e
l'idrogeno sono elettrofili, come mostrato nel disegno a destra. Per la
precisione, la natura bipolare del legame O-H è tale che gli alcoli sono acidi
molto più forti degli alcani (di circa 1030 volte), e quasi della
stessa misura, più forti degli eteri (alcani sostituiti dall'ossigeno, ma che
non hanno un gruppo O-H). La regione più reattiva in una molecola di alcool è
il gruppo ossidrile, a dispetto del fatto che la forza del legame O-H è
significativamente più alta dei legami C-C, C-H e C-O, sottolineando ancora una
volta la differenza tra stabilità termodinamica e stabilità chimica.
Per una discussione su come l'acidità sia influenzata dalla struttura molecolare cliccare qui. |
|---|
Sostituzione elettrofila sull'ossigeno |
|---|
A causa della sua esaltata acidità, l'atomo di idrogeno del gruppo ossidrile è rimpiazzabile abbastanza facilmente da altri sostituenti. Un semplice esempio è la facile reazione di alcoli semplici con il sodio (e con l'idruro di sodio), come descritto dalla prima equazione, appresso. Un'altra di tali reazioni di sostituzione è lo scambio isotopico che avviene miscelando un alcool con ossido di deuterio (acqua pesante). Questo scambio, catalizzato da un acido o da una base, è molto veloce sotto condizioni normali, poiché risulta difficile evitare tracce di questi catalizzatori nella maggior parte dei sistemi sperimentali.
2 R–O–H + 2 Na  2 R–O(–)Na(+) + H2 2 R–O(–)Na(+) + H2 |
R–O–H + D2O  R–O–D + D–O–H R–O–D + D–O–H |
Il meccanismo, grazie al quale hanno luogo molte reazioni di sostituzione di questo tipo, è semplice e diretto. L'atomo di ossigeno è nucleofilo e quindi propenso all'attacco degli elettrofili. Il risultante intermedio "-onio" cede allora un protone ad una base, dando il prodotto di sostituzione. Se non è presente un forte elettrofilo, la nucleofilia dell'ossigeno può essere aumentata per trasformazione nella sua base coniugata (un alcossido). Questo potente nucleofilo attacca allora il debole elettrofilo. Queste due varianti del meccanismo delle sostituzione sono illustrate nel diagramma seguente.

La preparazione dell'ipoclorito di tert-butile dall'alcool tert-butilico è un esempio di alogenazione elettrofila dell'ossigeno, ma questa reazione è confinata agli alcoli terziari in quanto gli ipocloriti primari e secondari perdono HCl per dare aldeidi e chetoni. Nella seguente equazione, Cl(+) può essere considerato come l'elettrofilo.
(CH3)3C–O–Cl + NaCl + H2O
La sostituzione alchilica del gruppo ossidrile conduce agli eteri. Questa reazione fornisce esempi sia di sostituzione elettrofila energica (prima equazione sotto), sia di sostituzione elettrofila debole (seconda equazione). L'ultima reazione SN2 è conosciuta come sintesi di eteri di Williamson, e viene generalmente usata soltanto con alogenuri alchilici primari poiché la forte base costituita dall'alcossido conduce ad eliminazione E2 degli alogenuri alchilici secondari e terziari.
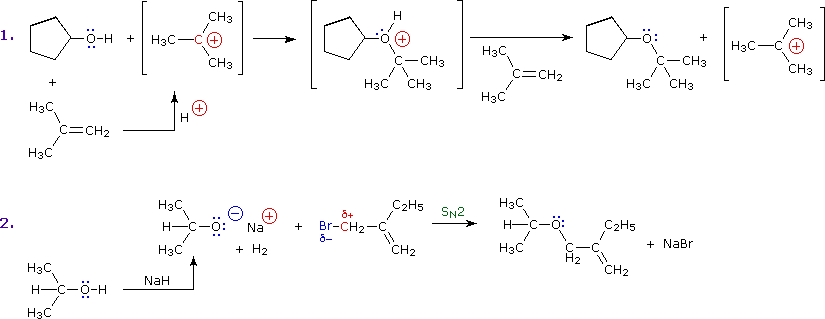
Una delle più importanti reazioni di sostituzione indirizzata all'ossigeno è la formazione di esteri derivante dalla reazione degli alcoli con derivati elettrofili degli acidi carbossilici e solfonici. La seguente illustrazione mostra le formule generiche di questi reagenti e dei loro esteri prodotti, in cui il gruppo R'–O– rappresenta la parte alcolica. L'atomo elettrofilo presente nei cloruri acilici e nelle anidridi è colorato in rosso. Esempi di specifiche reazioni di esterificazione si possono selezionare dal menu sotto al diagramma, che saranno visualizzate nella stessa finestra.